HIPERPLASIA PROSTÁTICA BENIGNA
Es un trastorno muy frecuente provocado por una hiperplasia epitelial y del estroma periuretral, que comprime la uretra; los síntomas se deben a la obstrucción del flujo urinario. Se encuentran características histológicas de HPB en el 20% de los hombres a los 4 años, en el 70% a los 60 y en el 90% a los 70 años; sólo la mitad de ellos tienen una hipertrofia prostática detectable clínicamente y, de ellos, sólo el 50% presenta síntomas. Aproximadamente el 30% de los hombres americanos de raza blanca meyores de 50 años sufren síntomas moderados a intensos.
PATOGENIA
El mediador fundamental de este proceso es la dihidrotestosterona (DHT), sintetizada por las células estromales prostáticas a partir de la testosterona circulante mediante la actividad de la 5 alfa-reductasa, de tipo 2.
- DHT se liga al receptor de andrógenos nuclear (RA) en las células epiteliales y estromales, lo que activa la transcripción de los genes dependientes de andrógenos.
- DHT no es un mitógeno directo, pero aumenta la producción de factores de crecimiento secundarios y sus receptores, sobre todo el factor de crecimiento fibroblástico-7 (FGF-7) por las células estromales.
- FGF-7 actúa de forma paracrina y estimula la proliferación de las células estromales, inhibiendo la apoptosis epitelial.
- la moyor producción de FGF-1 y FGF-2 y del factor de crecimiento transformante beta (TGF beta) participan también porque regulan la proliferación de los fibroblastos.
MORFOLOGÍA
 1.MACROSCÓPICA: la glándula aumenta de tamaño por la presencia de nódulos, que afectan principalmente a la zona transicional y periuretral; la superficie de corte muestra nódulos bien delimitados, que oscilan desde nódulos firmes y gris pálido (constituidos principalmente por estroma fibromuscular) a otros rosado-amarillentos blandos (de predominio glandular).
1.MACROSCÓPICA: la glándula aumenta de tamaño por la presencia de nódulos, que afectan principalmente a la zona transicional y periuretral; la superficie de corte muestra nódulos bien delimitados, que oscilan desde nódulos firmes y gris pálido (constituidos principalmente por estroma fibromuscular) a otros rosado-amarillentos blandos (de predominio glandular).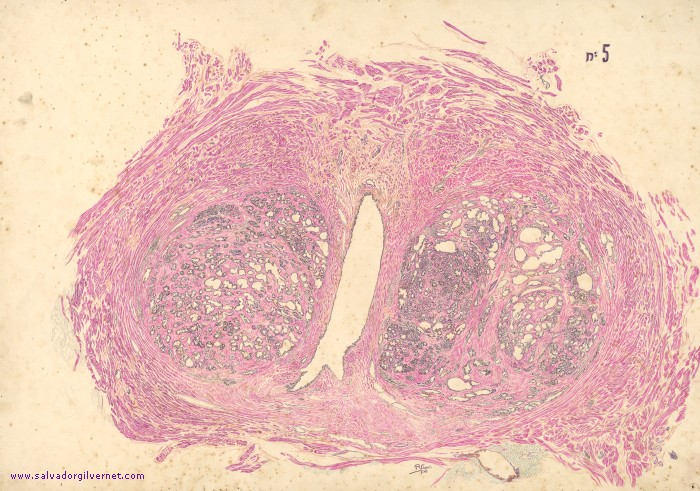
2. MICROSCÓPICA: los nódulos aparecen constituidos por mezclas variables de glándulas y estroma fibromuscular proliferativo; las glándulas se revisten por dos capas de células: una basal de epitelio cúbico bajo cubierta por otra de células cilíndricas secretoras. Ortos cambios son metaplasia escamosa e infartos.
CLÍNICA
Los síntomas de la obstrución de la vía urinaria se deben al aumento de tamaño de la próstata, la compresión extrínseca de la uretra y la contracción de la próstata mediada por el músculo liso. El consiguiente aumento de resistencia al flujo de orina es responsable de una hipertrofia vesical con distensión y retención urinaria. Los pacientes consultan por:
- Frecuencia urinaria, nicturia y dificultad para iniciar y detener el chorro de orina.
- Estasis urinaria crónica con el consiguiente sobrecrecimiento bacteriano e ITU.
- Divertículos vesicales e hidronefrosis.
El tratamiento incluye alfa-bloqueantes, que inhiben los receptores alfa1-adrenérgicos implicados en el tono del músculo liso prostático. Además, los inhibidores de la 5-alfa-reductasa pueden reducir la estimulación mediad por los DHT de base y, en los casos recalcitrantes, se puede optar por la reducción de volumen quirúrgica de la próstata.







